> CAS號(hào)數(shù)據(jù)庫(kù) > 102-54-5 > 102-54-5 / Trends in Chemistry綜述:二茂鐵不對(duì)稱C-H鍵功能化的機(jī)遇和挑戰(zhàn)
手機(jī)掃碼訪問(wèn)本站

微信咨詢
二茂鐵在有機(jī)合成、材料科學(xué)、生物醫(yī)學(xué)研究等領(lǐng)域發(fā)揮著重要作用。具有平面手性的二茂鐵尤其適合作為不對(duì)稱催化的配體或催化劑。因此,在二茂鐵主鏈上引入平面手性基團(tuán)有重大意義。中國(guó)科學(xué)院上海有機(jī)化學(xué)研究所游書(shū)力研究員團(tuán)隊(duì)近日在Cell Press旗下Trends in Chemistry期刊上發(fā)表綜述,闡述了二茂鐵對(duì)映選擇性C-H鍵功能化的最新進(jìn)展:即利用鈀、銥、銠、金、鉑和鎳催化C–H鍵功能化反應(yīng)合成平面手性二茂鐵。這些研究成果為平面手性二茂鐵的設(shè)計(jì)與合成提供了豐富的依據(jù)。
亮點(diǎn):
平面手性二茂鐵作為支架廣泛應(yīng)用于合成高效配體和催化劑;
過(guò)渡金屬直接催化C-H鍵官能化為合成平面手性二茂鐵提供了一種簡(jiǎn)單、方便、高效的方法。與非對(duì)映選擇鄰位導(dǎo)向金屬化反應(yīng)(DoM)、對(duì)映選擇DoM和手性拆分等傳統(tǒng)方法相比,該類方案具有顯著優(yōu)勢(shì);
適宜催化體系的發(fā)展使得從二茂鐵制備具有對(duì)映選擇性的C-C鍵體系成為可能。廉價(jià)金屬催化劑的發(fā)現(xiàn)進(jìn)一步提高了此類方案的實(shí)用性;
不對(duì)稱C-H鍵功能化得到的各種平面手性二茂鐵產(chǎn)物為設(shè)計(jì)和合成不對(duì)稱催化體系的高效配體和催化劑奠定了基礎(chǔ)。
近幾十年來(lái),人們對(duì)平面手性二茂鐵的不對(duì)稱合成進(jìn)行了廣泛研究。常用的策略包括非對(duì)映選擇臨位導(dǎo)向金屬化(DoM)、對(duì)映選擇性DoM和去對(duì)稱化。然而,這些方法往往依賴于預(yù)先設(shè)置的手性助劑或化學(xué)計(jì)量比的手性堿。由于多數(shù)情況下使用了金屬有機(jī)試劑,官能團(tuán)耐受性差和原子利用率低等問(wèn)題阻礙了這些方法的進(jìn)一步發(fā)展。相比之,催化不對(duì)稱合成反應(yīng)能夠更直接有效地合成平面手性二茂鐵結(jié)構(gòu)。
利用Pd(II)催化二茂鐵進(jìn)行不對(duì)稱C-H鍵功能化的主要研究如圖1所示。2013年,You團(tuán)隊(duì)利用空氣作為終端氧化劑,實(shí)現(xiàn)了二烷基氨基甲基二茂鐵衍生物與芳基硼酸的不對(duì)稱C–H芳基化反應(yīng)(圖1A). 并指出該反應(yīng)是在內(nèi)部堿協(xié)助下,通過(guò)金屬化-去質(zhì)子化協(xié)同作用(CMD)選擇性裂解二茂鐵1a中的C–H鍵而生成Pd(II)中間體A。然后,中間體A經(jīng)苯基硼酸金屬轉(zhuǎn)移反應(yīng)轉(zhuǎn)化為中間體B,再還原消除B生成產(chǎn)物3a,同時(shí)釋放Pd(0)物種及被空氣氧化生成Pd(II)物種,完成催化循環(huán)。同年,Wu和Cui團(tuán)隊(duì)分別使用類似策略,獨(dú)立報(bào)道了不對(duì)稱氧化Heck反應(yīng)(圖1B). 諸如丙烯酸酯、苯乙烯衍生物、乙烯基環(huán)己烷等各種烯烴都適合于該反應(yīng),并且產(chǎn)率高,對(duì)映選擇性好。You團(tuán)隊(duì)利用空氣為氧化劑,實(shí)現(xiàn)Pd催化的二烷基胺甲基二茂鐵與二芳基炔烴的不對(duì)稱C–H環(huán)化反應(yīng)(圖1C)。2014年,Wu、Cui等人開(kāi)發(fā)了鈀催化二烷基氨基甲基二茂鐵與1,2-二酮的對(duì)映選擇性C–H酰化反應(yīng)(圖1D). 2015年,You團(tuán)隊(duì)開(kāi)發(fā)了二茂鐵與雜芳烴的不對(duì)稱二重C–H氧化交叉偶聯(lián)反應(yīng)(圖1E),并在2019年進(jìn)一步研究發(fā)現(xiàn),在Pd(II)催化的二烷基甲基二茂鐵不對(duì)稱C-H/C-H交叉偶聯(lián)反應(yīng)中,惡唑和噻唑是合適的底物(圖1F). 2020年,Wu、Cui報(bào)道了在鈀催化鄰位烯基化反應(yīng)中,羧酸可作為弱配位導(dǎo)向基團(tuán),以合成平面手性1,2-二取代二茂鐵羧酸衍生物(圖1G). 2018年,Jin,Xu等人首次使用催化量的L-叔亮氨酸作為手性非穩(wěn)態(tài)導(dǎo)向基團(tuán)實(shí)現(xiàn)二茂鐵酮的鈀催化對(duì)映選擇性C–H芳基化反應(yīng)(圖1H)。反應(yīng)可能于底物10a和L-叔亮氨酸生成C開(kāi)始,經(jīng)去質(zhì)子化和金屬絡(luò)合后,通過(guò)CMD生成中間體D。D隨后與碘苯氧化加成生成Pd(IV)中間體E,經(jīng)還原消除得到芳基化物F,并釋放Pd(II)催化劑。最后,亞胺經(jīng)過(guò)水解步驟生成產(chǎn)物11a和L-叔亮氨酸。
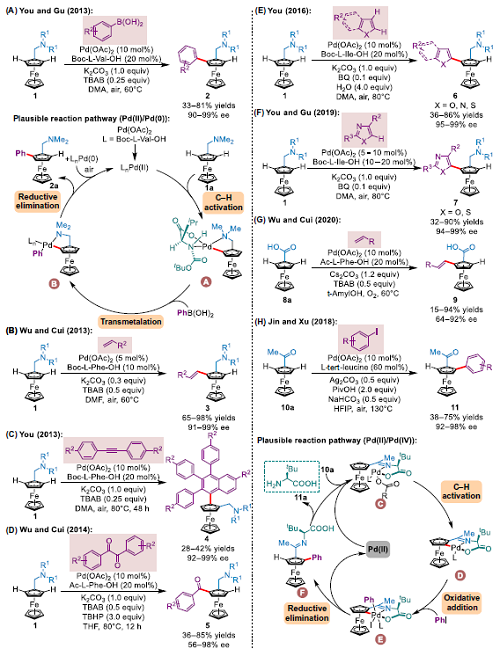
▲圖1 Pd(II)催化二茂鐵不對(duì)稱C-H鍵功能化
近年來(lái)研究工作證實(shí)Pd(0)催化分子內(nèi)C–H芳基化反應(yīng)是一種很有前景的平面手性二茂鐵合成方法。2014年,You團(tuán)隊(duì),Gu以及Kang團(tuán)隊(duì)分別通過(guò)對(duì)映選擇性分子內(nèi)C–H芳基化反應(yīng)合成了平面手性二茂鐵。You和Gu團(tuán)隊(duì)研究發(fā)現(xiàn),使用BINAP為配體,Pd催化2-溴代苯甲酰二茂鐵的對(duì)映選擇性環(huán)化反應(yīng)可有效地生成相應(yīng)的芳基化產(chǎn)物(圖3A)。在溫和條件下,二茂鐵衍生物的收率高達(dá)99%,目標(biāo)產(chǎn)物含量高達(dá)99%.該反應(yīng)的可能機(jī)理是:首先,Pd(0)催化劑與鹵化芳基12a通過(guò)氧化加成反應(yīng)生成芳基鈀中間體G,經(jīng)分子內(nèi)對(duì)映選擇性C–H活化反應(yīng)后,生成平面手性環(huán)鈀中間體H。H經(jīng)還原消除生成環(huán)化產(chǎn)物13a,同時(shí)Pd(0)催化劑再生。2014年,Liu,Zhao等人利用TADDOL衍生的亞磷酰胺配體,獲得了高產(chǎn)率且具有對(duì)映選擇性的產(chǎn)物(圖2B). 2015年,You和Gu團(tuán)隊(duì)通過(guò)鈀催化分子內(nèi)C–H芳基化反應(yīng),高效合成了平面手性二茂鐵吡啶衍生物(圖2C),并進(jìn)一步研究了合成平面手性二茂鐵分子內(nèi)C–H烯基化反應(yīng)(圖2D). 2018年,Duan團(tuán)隊(duì)發(fā)表了鈀催化二茂鐵硫化物不對(duì)稱C–H芳基化反應(yīng)的工作(圖2E),制備了一系列手性二茂鐵噻吩類化合物,產(chǎn)率高、對(duì)映選擇性好。同年,Zhu,Luo等人使用Pd催化對(duì)映選擇性亞氨基C–H鍵活化反應(yīng),獲得一類新的平面手性吡啶[3,4-b]二茂鐵(圖2F). 2020年,Liu團(tuán)隊(duì)開(kāi)發(fā)了一種高效的平面手性二茂鐵基吡咯烷酮合成方法,該方法利用BINOL衍生的手性磷酰胺為配體,通過(guò)鈀(0)催化,實(shí)現(xiàn)碳鈀化/不對(duì)稱C–H烯基化(圖2G).
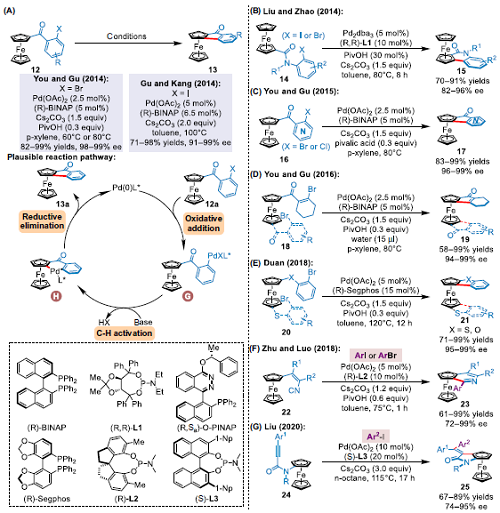
▲圖2Pd(0)催化二茂鐵不對(duì)稱C-H鍵功能化
2014年,Shibata團(tuán)隊(duì)使用手性二烯為配體,利用異喹啉-2-基導(dǎo)向基團(tuán)抑制二次烷基化反應(yīng),首次實(shí)現(xiàn)了Ir催化二茂鐵與烯烴的不對(duì)稱C–H烷基化反應(yīng)(圖3A),并初步提出催化循環(huán)策略。首先[Ir(coe)2Cl]2所產(chǎn)生的Ir和手性配體與26a的N原子配位,形成中間體I。通過(guò)氧化斷裂配體芳基的鄰位C–H加成金屬得到中間體J. 然后,將烯烴插入到Ir–H鍵中,得到物種K。最后K經(jīng)歷還原消除得到平面手性產(chǎn)物27a,同時(shí)釋放Ir(I)與底物配位生成的I進(jìn)行下一次催化循環(huán)。2015年,Shibata等人在脫氫偶聯(lián)反應(yīng)中利用手性二烯烴,獲得產(chǎn)率高和對(duì)映選擇性好的硅烷化產(chǎn)物(圖3B)。同時(shí),He和Murai/Takai分別獨(dú)立地證明手性雙膦配體可用于不對(duì)稱脫氫反應(yīng)硅烷化反應(yīng)。該反應(yīng)首先得到硅銠物種M,經(jīng)對(duì)映選擇性C–H裂解后生成平面手性中間體N,最終經(jīng)還原消除得到苯并硅基二茂鐵產(chǎn)物29,實(shí)現(xiàn)銠催化劑再生。2018年,Zhao團(tuán)隊(duì)開(kāi)發(fā)了一種2-二茂鐵基取代酚硅醚的不對(duì)稱脫氫C–H硅烷基化反應(yīng),合成了含六元硅環(huán)的對(duì)映體平面手性二茂鐵結(jié)構(gòu)(圖3C)。
2019年,You,Gu等人報(bào)道了Rh(I)催化硫代雙烯酮導(dǎo)向的二茂鐵對(duì)映選擇性C-H鍵芳基化反應(yīng)。使用商品化芳基碘為偶聯(lián)劑,生成產(chǎn)率高、對(duì)應(yīng)選擇性好的平面手性二茂鐵(圖3D). 在溫和的反應(yīng)條件下,使用芳基鹵化物,利用對(duì)映選擇性Rh(I)催化吡啶,實(shí)現(xiàn)二茂鐵C–H芳基化反應(yīng),高效制備了平面手性二茂鐵(圖3E). 該反應(yīng)過(guò)程首先是含有單磷酸酯配體的Rh(I)催化劑與26a的N原子配位,形成絡(luò)合物O。通過(guò)叔丁醇輔助脫質(zhì)子化進(jìn)行可逆金屬環(huán)化加成反應(yīng)得到中間產(chǎn)物P。然后,氧化加成芳基鹵化物與中間體P得到Q物質(zhì)。隨后,Q還原消除得到平面手性二茂鐵34a,同時(shí)釋放Rh(I)催化劑完成催化循環(huán)。
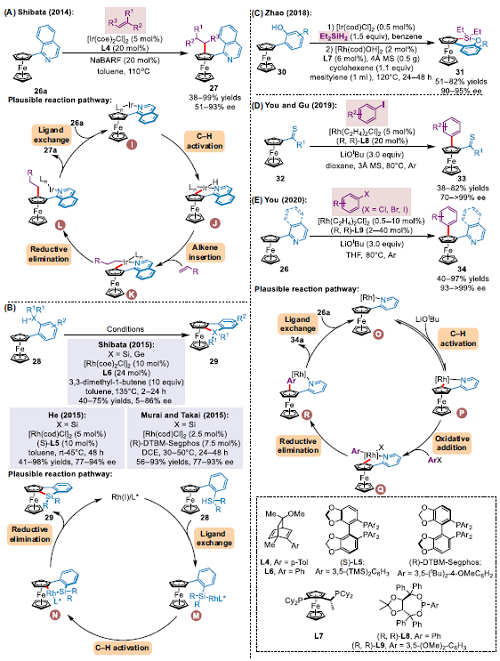
▲圖3 Ir/Rh催化二茂鐵不對(duì)稱C-H鍵功能化
過(guò)渡金屬催化分子內(nèi)環(huán)異構(gòu)化是合成菲和螺旋烯的一種有效方法。Urbano和Carreño開(kāi)發(fā)了一種用于合成芳香族三環(huán)二茂鐵的對(duì)映選擇性Au(I)催化環(huán)異構(gòu)化反應(yīng)(圖4A). 同年,Shibata團(tuán)隊(duì)使用類似方案,用Pt/(S,S)-Ph-BPE為催化系統(tǒng),合成平面手性二茂鐵(圖4B). 2019年,Shibata和Ito利用類似的策略,使用手性陽(yáng)離子Pt催化劑實(shí)現(xiàn)了對(duì)映選擇性合成含氮卓環(huán)平面手性二茂鐵的構(gòu)建(圖4C).
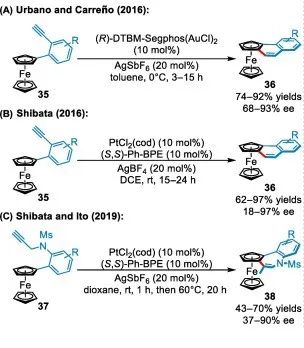
▲圖4 Pt/Au催化二茂鐵不對(duì)稱C-H鍵功能化
過(guò)去幾年對(duì)Ni催化C–H活化反應(yīng)開(kāi)展了廣泛研究,但鮮有關(guān)于平面手性結(jié)構(gòu)的報(bào)道。最近,Ye團(tuán)隊(duì)首次利用Ni催化劑催化雙重C–H鍵和炔烴發(fā)生不對(duì)稱氧化環(huán)化,提出了一種高效合成平面手性二茂鐵酰胺衍生物的方法,并證明了大尺寸BINOL二苯醇衍生的SPO使Ni-Al共催化劑對(duì)反應(yīng)起關(guān)鍵作用(圖5). 作者提出該反應(yīng)由Ni和Al同時(shí)與雙功能SPO發(fā)生協(xié)同作用。之后,原位形成的催化劑與底物配位活化C–H鍵,生成中間體V。隨后,炔烴插入,所生成的中間體經(jīng)過(guò)還原消除生成副產(chǎn)物41,或發(fā)生二次C–H激活,通過(guò)SEAr機(jī)制生成中間體X。炔烴第二次插入中間體X和隨后的還原消除反應(yīng)最終生成目標(biāo)產(chǎn)物40a.。
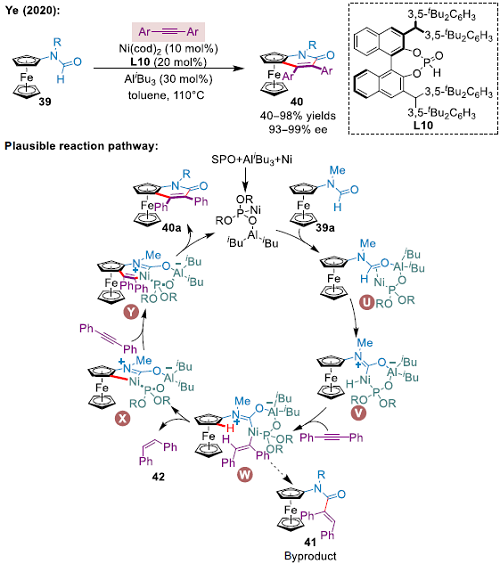
▲圖5 Ni催化二茂鐵不對(duì)稱C–H鍵功能化反應(yīng)
過(guò)渡金屬催化對(duì)映選擇性C-H官能化反應(yīng)是一種直接、便捷、高效的合成平面手性二茂鐵的方案。本文中綜述了二茂鐵直接不對(duì)稱C-H鍵功能化的最新進(jìn)展。與傳統(tǒng)的方法相比,這些方法在構(gòu)建平面手性分子時(shí)更具優(yōu)勢(shì)。目前為止,盡管取得了可嘉成果,但這一領(lǐng)域仍處于初級(jí)探索階段,面臨諸多問(wèn)題。例如,二茂鐵對(duì)映選擇性C-H鍵官能化還不是合成手性配體和催化劑的常用策略;如何利用這些新方法設(shè)計(jì)更高效的平面手性二茂鐵配體和催化劑;二茂鐵的高對(duì)映選擇性C-H官能化形成碳雜原子鍵(如C-N、C-S和C-P)的報(bào)道很少;高區(qū)域選擇性二茂鐵的合成,如1,3-二取代或1,1’-二取代二茂鐵,也是該領(lǐng)域的一個(gè)發(fā)展機(jī)遇。此外,反應(yīng)的進(jìn)行通常需要高的催化劑負(fù)載量,這會(huì)阻礙其進(jìn)一步應(yīng)用。為應(yīng)對(duì)這些挑戰(zhàn),利用對(duì)映選擇性C–H官能化更有效地合成平面手性二茂鐵的研究會(huì)持續(xù)進(jìn)行,更具實(shí)用性的催化體系終將會(huì)解決問(wèn)題并實(shí)現(xiàn)應(yīng)用。
Asymmetric C–H Bond Functionalization of Ferrocenes: New Opportunities and Challenges